Un exemple d’Application Scientifique
d’APL
: La Simulation des Spectres de
RÉSONANCE MAGNÉTIQUE
par Claude Chachaty
ou
:
Le rôle pratique d'APL
pour l'écriture de programmes efficaces
L’auteur de cet article, Claude Chachaty,
n’utilise couramment, depuis plus de quinze ans, qu’un seul langage de
programmation pour développer des logiciels dans toutes ses activités de
recherche : APL.
C. Chachaty, après avoir pris récemment sa
retraite, continue à publier dans la communauté scientifique mondiale les
résultats d’expériences qu’il a accumulés durant de longues années dans ce qui
était, à l’époque où il en assurait la direction, le second laboratoire de
recherche en RMN en France, de par l’importance de ses installations (C. C. est
l’auteur d’environ 180 publications dans de grandes revues internationales).
Cet article traite d’un sujet difficile,
exigeant un très haut niveau mathématique et une bonne connaissance dans de
nombreux domaines de la physique. Le but poursuivi par AFAPL en publiant cet
article, est de montrer au lecteur que le champ d’application d’APL est plus
vaste que ce que l’on croit, et peut faire progresser la science dans des
domaines où chacun doit se sentir concerné, car les techniques de RPE
(Résonance Paramagnétique Electronique) et RMN (Résonance Magnétique Nucléaire)
ont et auront encore des retombées considérables notamment dans la pratique
médicale. Les possibilités considérables offerte par APL en recherche
fondamentale sont peu exploitées ou tout
simplement ignorées par les universitaires, aussi bien en France qu'à
l'étranger. Au cours de ces quinze dernières années, l'auteur de cet article a
utilisé intensivement APL dans ses recherches et contribué à faire connaître
les applications de ce langage dans le
domaine de la spectroscopie de résonance
magnétique [1-3], qu'il continue à développer.
La résonance magnétique comporte deux
branches principales, la résonance paramagnétique électronique (RPE) et la
résonance magnétique nucléaire (RMN) respectivement découvertes en URSS par
Zavoisky [4] et aux Etats-Unis par Bloch [5] et Purcell [6].
La RMN est bien connue du grand
public en raison de ses applications médicales (scanner). Le principe de la
résonance magnétique peut être résumé de la manières suivante : les électrons
et la plupart des isotopes nucléaires possèdent un moment angulaire de spin que
l'on représente classiquement comme un dipôle magnétique engendré par la
rotation d'une charge électrique. Dans un champ magnétique ![]() , les spins
effectuent un mouvement de précession de pulsation
, les spins
effectuent un mouvement de précession de pulsation ![]() autour de ce champ. Prenons l'exemple d'un spin électronique;
pour un nombre quantique de spin S, il existe 2S+1 valeurs possibles pour sa
projection sur la direction
autour de ce champ. Prenons l'exemple d'un spin électronique;
pour un nombre quantique de spin S, il existe 2S+1 valeurs possibles pour sa
projection sur la direction ![]() soit
soit ![]() en unité
en unité ![]() , h étant la
constante de Planck. Il en est de même pour un spin nucléaire I. A chaque valeur de
, h étant la
constante de Planck. Il en est de même pour un spin nucléaire I. A chaque valeur de ![]() (ou de
(ou de ![]() ) correspond un niveau d'énergie. Les
transitions permises entre ces niveaux fondamentaux (résonances) sont induites
par la composante magnétique
) correspond un niveau d'énergie. Les
transitions permises entre ces niveaux fondamentaux (résonances) sont induites
par la composante magnétique ![]() d'un rayonnement électromagnétique de
pulsation
d'un rayonnement électromagnétique de
pulsation ![]() , polarisé perpendiculairement à
, polarisé perpendiculairement à ![]() . Actuellement, les fréquences des
spectromètres se situent entre 1
GHz et 250 GHz pour la RPE et entre 60 et 750 MHz pour la RMN c'est-à-dire
respectivement dans le domaine des hyperfréquences ou de l'infrarouge lointain
et dans celui des radiofréquences. En l’absence de toute interaction autre que
celle entre les spins et les champs magnétiques
. Actuellement, les fréquences des
spectromètres se situent entre 1
GHz et 250 GHz pour la RPE et entre 60 et 750 MHz pour la RMN c'est-à-dire
respectivement dans le domaine des hyperfréquences ou de l'infrarouge lointain
et dans celui des radiofréquences. En l’absence de toute interaction autre que
celle entre les spins et les champs magnétiques ![]() , la résonance
électronique ou nucléaire serait observable pour
, la résonance
électronique ou nucléaire serait observable pour ![]() ,
, ![]() étant l'intensité du champ magnétique et
étant l'intensité du champ magnétique et ![]() , une constante
caractéristique de l'électron libre ou d'un noyau donné. En réalité, les spins
sont soumis à de multiples interactions
provenant en particulier des spins électroniques ou nucléaires voisins, ce qui
a pour effet de diviser les niveaux fondamentaux en sous-niveaux, ceux-ci étant
modulés par les fluctuations de champs magnétiques ou électriques locaux. C'est
pourquoi les spectres de résonance comportent en général plusieurs raies et que
ces raies ont une largeur finie.
, une constante
caractéristique de l'électron libre ou d'un noyau donné. En réalité, les spins
sont soumis à de multiples interactions
provenant en particulier des spins électroniques ou nucléaires voisins, ce qui
a pour effet de diviser les niveaux fondamentaux en sous-niveaux, ceux-ci étant
modulés par les fluctuations de champs magnétiques ou électriques locaux. C'est
pourquoi les spectres de résonance comportent en général plusieurs raies et que
ces raies ont une largeur finie.
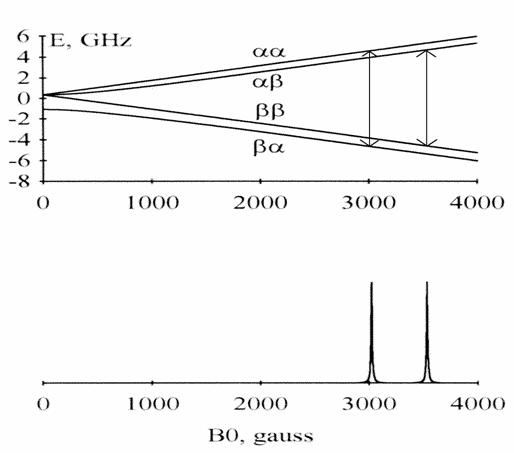
Figure 1. Niveaux d'énergie et spectre
d'absorption de RPE de l'atome H à la fréquence de 9,3 GHz. Les états
![]() sont représentés par
sont représentés par ![]() ,
, ![]() correspondant à
correspondant à ![]() et
et ![]() à
à ![]() . La règle de sélec-tion
. La règle de sélec-tion ![]() n'autorise que les transitions
n'autorise que les transitions ![]() .
.
Un exemple simple d’interaction entre
deux spins 1/2, ceux de l'électron et du proton, est donné par l'atome
d'hydrogène, facilement observable dans l'hydrogène gazeux soumis à une
décharge micro-onde ou dans des verres minéraux irradiés à basses températures.
Un spectre analogue à celui de la figure 1 serait obtenu en RMN dans un système
à deux spins 1/2 comme l'acide fluorhydrique HF, si l'on observe soit le fluor,
soit le proton. La différence essentielle entre la RPE et la RMN est dans la nature des espèces chimiques que
permettent d'étudier ces deux spectroscopies. La RMN concerne plutôt les
molécules stables, généralement diamagnétiques, et son usage est beaucoup plus
général. C'est une méthode privilégiée d'identification et d'étude
conformationnelle des molécules dans les liquides et les solides. En
collaboration avec G. Langlet, nous avons développé des logiciels APL pour
l'étude par RMN des conformations et de la dynamique de molécules flexibles en
milieu fluide [1]. La RPE
dont il sera question uniquement par la suite, ne concerne par contre que les
molécules paramagnétiques possédant au moins un électron non apparié,
c'est-à-dire les radicaux provenant de la fragmentation ou de l'ionisation
d'une molécule, certains complexes métalliques ainsi que l'oxygène qui est un
biradical. Ces molécules, généralement très réactives, sont responsables en
particulier des réactions de réduction, d'oxydation et de polymérisation.
L'analyse d'un spectre de RPE donne trois
types d'informations sur une molécule paramagnétique :
- Nature, conformation, structure
électronique, d'après les positions et les intensités
relatives des raies.
- Mobilité rotationnelle et
translationnelle, d'après l'évolution des positions et des
largeurs de raies avec la température.
- Concentration dont l'évolution
éventuelle en fonction du temps donne la cinétique de
réaction.
Les deux premiers types d'informations
nécessitent généralement des simulations de spectres en essayant plusieurs jeux de paramètres : les
valeurs principales du tenseur gyromagnétique (g) et les largeurs de raies corres-pondantes, celles des tenseurs
de couplage hyperfin (A) ainsi que
les orientations relatives des axes principaux de ces tenseurs. g et A correspondent respectivement à l’interaction du spin électronique
avec le champ ![]() et avec un spin nucléaires. Ces tenseurs sont
représentés par des matrices symétriques
et avec un spin nucléaires. Ces tenseurs sont
représentés par des matrices symétriques ![]() dont les valeurs propres sont de la forme
dont les valeurs propres sont de la forme ![]() avec u = x, y ou z.
avec u = x, y ou z.
Pour une distribution sphérique du spin
électronique autour d'un noyau (orbitale s) comme dans le cas de l'atome
H, les termes anisotropes ![]() disparaissent et l'on obtient un spectre
indépendant de l'orientation du champ magnétique. Dans la majorité des cas
cependant, la distribution de la densité de spin électronique est anisotrope de
sorte que les positions et les écarts des raies dépendent de l'orientation des
axes x,y et z par rapport à
disparaissent et l'on obtient un spectre
indépendant de l'orientation du champ magnétique. Dans la majorité des cas
cependant, la distribution de la densité de spin électronique est anisotrope de
sorte que les positions et les écarts des raies dépendent de l'orientation des
axes x,y et z par rapport à ![]() . Si le milieu étudié
est une matrice rigide amorphe ou polycristalline, le signal RPE est une somme
pondérée d'une quasi-infinité de spectres correspondant à toutes les
orientations de
. Si le milieu étudié
est une matrice rigide amorphe ou polycristalline, le signal RPE est une somme
pondérée d'une quasi-infinité de spectres correspondant à toutes les
orientations de ![]() définies par les angles polaires
définies par les angles polaires ![]() dans un référentiel moléculaire. Il en résulte
des formes de raies assez complexes comme on peut en juger en comparant les
figures 1 et 2 qui correspondent toutes deux au couplage du spin électronique
avec un seul proton.
dans un référentiel moléculaire. Il en résulte
des formes de raies assez complexes comme on peut en juger en comparant les
figures 1 et 2 qui correspondent toutes deux au couplage du spin électronique
avec un seul proton.
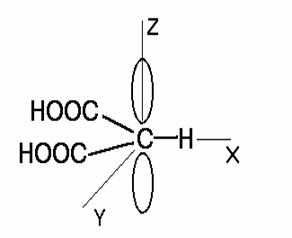
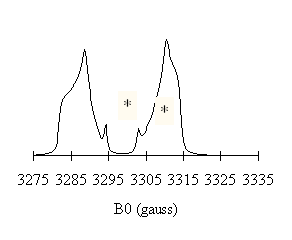
Figure 2. Spectre
d'absorption du radical malonyle à 9.3 GHz calculé d'après les données de
McConnell et al. , J. Am. Chem. Soc. 82,
766 (1960). Les raies marquées (*) correspondent aux transitions
"interdites" ![]() . La direction Z est l'axe de l'orbitale de l'électron non
apparié.
. La direction Z est l'axe de l'orbitale de l'électron non
apparié.
Les spectres de RPE
sont enregistrés à fréquence ![]() fixe et à champ
fixe et à champ ![]() variable, généralement
sous forme de dérivée première d'absorption, et le programme de simulation
procède par les étapes suivantes :
variable, généralement
sous forme de dérivée première d'absorption, et le programme de simulation
procède par les étapes suivantes :
1 - Paramètres
Invariants : fréquence du spectromètre, domaine spectral, valeurs du spin électronique et des spins nucléaires.
Ajustables :
valeurs principales des tenseurs A , g et D (interaction
dipolaire entre spins électroniques pour ![]() ), largeurs de raies lorentziennes ou gaussiennes.
), largeurs de raies lorentziennes ou gaussiennes.
2 - Dépendance angulaire des probabilités de transition ![]() et des largeurs
de raies
et des largeurs
de raies ![]() .
.
3 - Calcul des champs de résonance ![]() .
.
4 - Pour chaque transition, sommation des spectres sur toutes les orientations suivant l'expression :
![]()
où F est la fonction de forme et N un facteur de normalisation, ce qui
donne en APL2, pour une forme lorentzienne[1] de
raie
(d1 d2 d3)„½¨B0
th phi
U„B0°.-,Br ª F„÷‘U×1+U×U„U÷‘U„(d1½1)°.×,sigma
S„S÷+/S„F+.×,P×(1±th)°.×d3½1
S
peut être ensuite convolué par une gaussienne et dérivé numériquement.
5 - Visualisation des spectres
expérimental et calculé.
6 - Si le résultat n'est pas satisfaisant,
retour en 1 pour ajuster les valeurs prin- cipales des tenseurs et les largeurs
de raie. Cette étape peut être automatisée à l'aide d'un programme
d'optimisation qui minimise la variance entre le spectre expérimental et le
spectre calculé.
La programmation en APL de ce calcul,
dont la figure 3 donne un exemple, est très simple. Le seul problème est celui
de la dimension des tableaux qu'il faut parfois fragmenter pour éviter WS FULL.

Figure 3. Dérivée première du
spectre expérimental et calculé (- - - - -) de l'ion vanadyle ![]() en matrice rigide de polymère
en matrice rigide de polymère ![]() .
Durée du calcul : 10 s en APL*PLUS II sur PC 486, 66 MHz, pour 530 valeurs de
.
Durée du calcul : 10 s en APL*PLUS II sur PC 486, 66 MHz, pour 530 valeurs de ![]() et 120 valeurs de q.
et 120 valeurs de q.
L'application majeure de la
RPE est actuellement la technique des sondes paramagnétiques ou marqueurs de
spin, généralement des radicaux stables nitroxydes, dont la mobilité est une
mesure de la viscosité locale du milieu étudié ou de la flexibilité d'une
macromolécule. On peut utiliser par exemple cette technique pour étudier
l'effet d'un plastifiant sur un polymère [7] ou l'adsorption d'un solvant dans des solides
poreux [8].
Cette mobilité est
caractérisée dans le cas le plus simple considéré ici, par un temps de
corrélation de réorientation ![]() qui correspond à peu près à la durée d'une rotation
aléatoire d'amplitude quadratique moyenne 30°. La forme des spectres et les
méthodes de simulation sont radicalement différentes selon l'ordre de grandeur de
qui correspond à peu près à la durée d'une rotation
aléatoire d'amplitude quadratique moyenne 30°. La forme des spectres et les
méthodes de simulation sont radicalement différentes selon l'ordre de grandeur de ![]() par rapport aux anisotropies des tenseurs A et g exprimées en fréquences. Si la réorientation est suffisamment
rapide, l'anisotropie de ces tenseurs est annulée en moyenne et l'on obtient
des raies fines et symétriques dont les largeurs augmentent linéairement avec
par rapport aux anisotropies des tenseurs A et g exprimées en fréquences. Si la réorientation est suffisamment
rapide, l'anisotropie de ces tenseurs est annulée en moyenne et l'on obtient
des raies fines et symétriques dont les largeurs augmentent linéairement avec ![]() .
Si la réorientation est lente, les écarts entre les raies sont plus grands que
dans le cas précédent et les raies sont larges et asymétriques. Enfin si la
réorientation est ultralente, le spectre est identique à celui observable en
milieu rigide. Pour un radical nitroxyde, à une fréquence de 9 GHz, la
frontière entre les domaines de réorientation rapide et de réorientation lente
se situe à
.
Si la réorientation est lente, les écarts entre les raies sont plus grands que
dans le cas précédent et les raies sont larges et asymétriques. Enfin si la
réorientation est ultralente, le spectre est identique à celui observable en
milieu rigide. Pour un radical nitroxyde, à une fréquence de 9 GHz, la
frontière entre les domaines de réorientation rapide et de réorientation lente
se situe à ![]() 2 10-9 s et la limite rigide vers 10-6 s.
2 10-9 s et la limite rigide vers 10-6 s.



Figure 4. Spectres RPE
expérimentaux et calculés (- - - - ) d'un radical nitroxyde phosphoré, en
milieux ( A ) rigide, ( B ) visqueux (![]() = 2.3 10-8 s), ( C ) fluide (
= 2.3 10-8 s), ( C ) fluide (![]() = 8.7 10-10 s). Leur
structure hyperfine provient de l'azote
(I = 1) et du phosphore (I = 1/2). Ces spectres ont été enregistrés au
laboratoire de P. Tordo à l'Université d'Aix-Marseille où a été synthétisé
le radical.
= 8.7 10-10 s). Leur
structure hyperfine provient de l'azote
(I = 1) et du phosphore (I = 1/2). Ces spectres ont été enregistrés au
laboratoire de P. Tordo à l'Université d'Aix-Marseille où a été synthétisé
le radical.
La figure 4 donne des exemples
de spectres expérimentaux et calculés pour ces trois domaines. Pour deux
d'entre eux, le temps de corrélation de réorientation a été obtenu par
simulation des spectres avec ajustement automatique de 3 à 5 paramètres, dont
![]() ,
à l'aide d'une fonction APL basée sur l'algorithme de Marquardt [9]
qui minimise les moindres carrés des écarts entre le spectre expérimental et le
spectre calculé. Le principe de ces
simulations est donné dans la réf. [8].
,
à l'aide d'une fonction APL basée sur l'algorithme de Marquardt [9]
qui minimise les moindres carrés des écarts entre le spectre expérimental et le
spectre calculé. Le principe de ces
simulations est donné dans la réf. [8].
La RPE, comme la RMN, a des
applications considérables en biologie, en particulier pour l'étude des
membranes [10] constituées en général par des doubles couches de
phospholipides. Les phospholipides comportent une tête polaire contenant un
groupe phosphate et deux longues chaînes hydrocarbonées. Les chaînes
hydrocarbonées constituent l'intérieur de la membrane et les têtes polaires se
placent aux interfaces avec l'eau. Les membranes biologiques sont en fait des
cristaux liquides dans lesquels les molécules fluctuent autour d'une direction
perpendiculaire aux interfaces. La fluidité d'une membrane varie entre les
interfaces et la partie médiane hydrocabonée. L'ordre moléculaire et la
fluidité peuvent être mesurées par RPE en incorporant une sonde radicalaire
nitroxyde de structure appropriée dans la membrane. On peut ainsi étudier
l'effet d'additifs comme le cholestérol
ou certains médicaments marqués au nitroxyde sur ces facteurs. Plusieurs programmes en APL ont été écrits
pour simuler les spectres de sondes paramagnétiques dans des cristaux liquides.
Ces programmes font appel à l'algorithme de Marquardt pour ajuster
automatiquement les paramètres de ces simulations, en particulier les temps de
corrélation de réorientation, les paramètres d'ordre qui donnent le degré
d'orientation moléculaire, éventuellement les vitesses d'échange entre deux
phases.

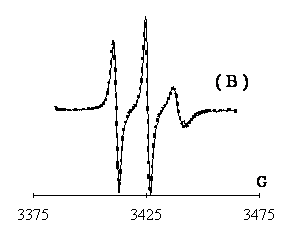
(A) : 5 NS,
![]() ,
, ![]() = 1.25 10-9
s. (B) : 16 NS,
= 1.25 10-9
s. (B) : 16 NS, ![]() ,
, ![]() = 8 10-10
s.
= 8 10-10
s.
La figure 5 permet de
comparer les spectres de deux sondes où le groupe nitroxyde se place soit au
voisinage de l'interface de la membrane avec l'eau (5 NS ), soit dans la
partie médiane de celle-ci (16 NS) . Les
paramètres d'ordre ![]() et les temps de corrélation
et les temps de corrélation ![]() mesurés sur ces deux sondes sont
caractéristiques d'un fort gradient de fluidité entre la surface et le centre
d'une double couche de phospholipide.
mesurés sur ces deux sondes sont
caractéristiques d'un fort gradient de fluidité entre la surface et le centre
d'une double couche de phospholipide.
Le logiciel de RPE dont il
est question ici ainsi que celui de RMN ont été initialement écrits en APL2
d'IBM sous VM. Ces logiciels existent actuellement en versions simplifiée pour
APL*PLUS, complète pour APL*PLUS II et en cours d'adaptation pour APL*PLUS III.
Nous n'avons pas relevé de différences notables entre les performances de ces
deux dernières versions, APL*PLUS III étant seulement un peu plus facile à
manipuler par un utilisateur ne connaissant pas APL.
REFERENCES
1 - C. Chachaty, G. Langlet, J. Chim.
Phys. 62, 613 (1985).
2 - C. Chachaty, ibid. 62, 621 (1985).
3 - C. Chachaty dans "Logiciels pour la Chimie", Société
Chimique de France, Paris (1991).
4 - E. Zavoisky, J. Phys. USSR, 9, 211 (1945).
5 - F.
Bloch, W. Hansen, M.E. Packard, Phys. Rev. 69, 127 (1946).
6 -
E.M. Purcell, H.C. Torrey, R.V. Pound, Phys. Rev. 69, 37 (1946).
7 - B.
Ranby, J.F. Rabek, "ESR Spectroscopy
in Polymer Research", chap. 11,
8 - C.
Chachaty, J. Chim.
Phys. 91, 1848 (1994).
9 - D.W. Marquardt, J. Soc. Ind.
Appl.
Math., 11, 431 (1963).
10-
"Biological Magnetic Resonance, 8 Spin Labeling , Theory and
Applications" édité par L.J. Berliner et J. Reuben, Plenum Press,
11- C. Wolf, C. Chachaty, Communication
au « 17th International EPR Symposium », Denver (1994).
________________
NdlR. Les graphiques de cet article ont
été réalisés à partir de calculs effectués en APL*PLUS III, grâce à l’interface
avec Excel Version 4.