PROGRAMMES
POUR LES APPLICATIONS PHYSICO-CHIMIQUES DE
CLAUDE CHACHATY
���� Un ensemble de programmes de simulation a
�t� �crit en langage APL pour de multiples applications de la r�sonance
magn�tique � la physico-chimie de la mati�re condens�e : phase liquide,
milieux vitreux ou polycristallins et cristaux liquides Ils concernent la
r�sonance paramagn�tique �lectronique (RPE) (1,2) ainsi que la
r�sonance et la relaxation magn�tique nucl�aires (RMN)(1,3,4)
dans les domaines o� ces techniques sont compl�mentaires de
���� Ces programmes ont pour but d�aider � l�analyse
des spectres et de d�terminer les param�tres spectroscopiques (tenseurs
magn�tiques) et dynamiques (temps de corr�lation de r�orientation) que l�on
peut en extraire. Ils s�appliquent aux domaines suivants
-Spectres
de radicaux, �tats triplets et biradicaux, ions paramagn�tiques de spin �lectronique
1/2 (RPE) ou de n�importe quel noyau (RMN) en matrices rigides pour d�terminer
les param�tres spectroscopiques.
-Spectres
de radicaux et biradicaux en phase liquide avec calcul des largeurs de raies en
fonction des temps de corr�lation de r�orientation ainsi que de l��change entre
spins �lectroniques.
-Temps
de relaxation ![]() �et
�et ![]() �nucl�aires en fonction
des temps de corr�lation de r�orientation et des mouvements intramol�culaires
dans les liquides et les cristaux liquides.
�nucl�aires en fonction
des temps de corr�lation de r�orientation et des mouvements intramol�culaires
dans les liquides et les cristaux liquides.
-Spectres
RPE de sondes paramagn�tiques� nitroxydes
et spectres de RMN de 2H,
��� Pour la plupart ces programmes pr�sentent deux
options :
-Simple
simulation pour �tudier l�effet de diff�rents param�tres sur la forme d�un
spectre et faire une �valuation pr�liminaire de leurs valeurs par comparaison
avec le spectre exp�rimental.
-Optimisation
des param�tres avec ajustement automatis� du spectre simul� au spectre exp�rimental
en utilisant l�algorithme de Levenberg-Marquardt(5).
���� Il existe
plusieurs zones non d�crites ici comportant des programmes utilitaires :
fonctions graphiques, transform�e de Fourier, d�convolution de spectres, �change
de fichiers avec d�autres applications Windows etc�
��� Toutes les
zones de travail existent en deux versions : APL2 (IBM) et APL+WIN
(APL2000) et sont disponibles sur demande aupr�s de l�auteur qui assure l�aide
scientifique et technique en particulier pour modifier un programme en vue
d�une nouvelle application. Le logiciel�
APL lui-m�me ne peut �tre fourni avec les logiciels de RPE et de RMN. On
peut se le procurer aupr�s d�IBM (www.ibm.com/software/ad/apl), d�APL2000 (www.apl2000.com) ou d�un fournisseur local.
��� Le choix d�APL comme langage de
programmation �est motiv� par les raisons
suivantes :
-Bien adapt� � l�alg�bre matricielle
(APL = Array Programming Language) tr�s utilis�e en r�sonance magn�tique.
-Programmation simplifi�e par l�existence
d�une vari�t� d�op�rateurs.
-Fonctionne en mode interactif
permettant une comparaison rapide entre les donn�es exp�rimentales et les
donn�es calcul�es avec un acc�s facile aux param�tres � modifier.
-Messages d�erreur tr�s explicites
permettant de poursuivre un calcul apr�s correction.
�� L�utilisation des programmes n�cessite
seulement� de conna�tre quelques symboles
APL.
1 � R�sonance Paramagn�tique Electronique.
HRESOL - Spectres � haute r�solution de
radicaux en milieu fluide avec couplage hyperfin (CHF) de plusieurs noyaux de
spin quelconque, avec prise en compte du d�placement de second ordre des raies
de r�sonance� et de la d�pendance de leur
largeur en fonction du nombre quantique magn�tique 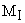 du noyau pr�sentant le plus grand couplage hyperfin.
du noyau pr�sentant le plus grand couplage hyperfin.
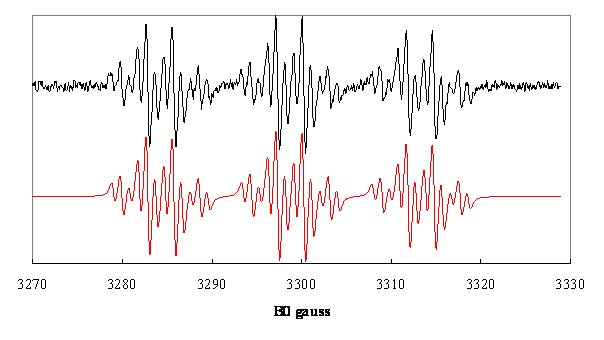
Figure 1. Spectres
exp�rimental �et simul� du radical ![]() �dans le m�thylt�trahydrofuranne
irradi� par γ � 77 K puis r�chauff� � 295 K .Les valeurs optimis�es des constantes de couplage hyperfin sont
�dans le m�thylt�trahydrofuranne
irradi� par γ � 77 K puis r�chauff� � 295 K .Les valeurs optimis�es des constantes de couplage hyperfin sont ![]() .
.
RIGMAT- Concerne les spectres de radicaux ou ions de spin �lectronique� S = 1/2� en matrice rigide vitreuse ou polycristalline dans les cas suivants:
1
� Anisotropie du tenseur spectroscopique�
g� sans couplage hyperfin.
2
� Anisotropie du tenseur g et du ou
des tenseur(s) de couplage hyperfin A
de 1 ou 2 noyaux avec d�placement du second ordre des raies de r�sonance, les
axes principaux de ces tenseurs �tant suppos�s communs.
Dans ces deux cas les valeurs principales optimis�es des tenseurs g et A peuvent �tre obtenues par simulation automatis�e des spectres.
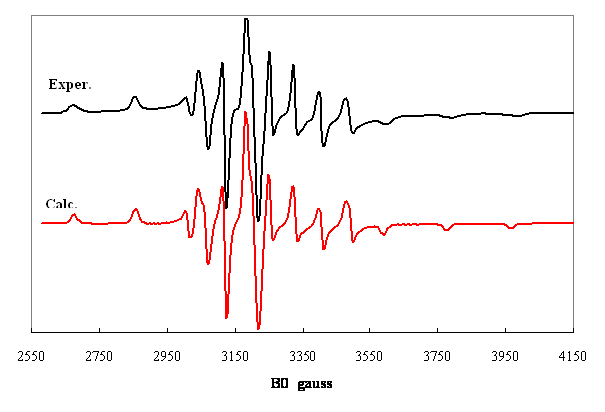
Figure 2. Spectre de l�ion ![]() �dans un gel r�ticul� de
poly(4-vinyl pyridine) � 250 K.
�dans un gel r�ticul� de
poly(4-vinyl pyridine) � 250 K.
Valeurs principales optimis�es des tenseurs A et g :
![]() , .
, .![]() .
.
L�axe Z est dirig� selon la liaison V-O.
3 � Orientations quelconques dans un r�f�rentiel
mol�culaire du tenseur g et des
tenseurs de couplage hyperfin de plusieurs noyaux de spins quelconques. Pour
les noyaux de spin I = 1/2� on prend en
compte les raies satellites r�sultant des transitions �� interdites �� ![]() ,
, ![]() . Les simulations de spectres sont bas�es sur le traitement
par perturbation du second ordre de l�Hamiltonien de spin par Iwasaki(6).
. Les simulations de spectres sont bas�es sur le traitement
par perturbation du second ordre de l�Hamiltonien de spin par Iwasaki(6).
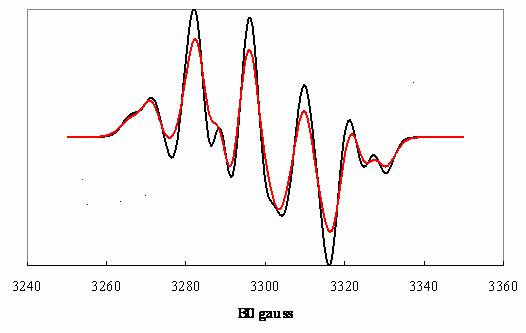
Figure 3.� Spectre du radical allyle ![]() �polyorient�
�polyorient�
: transitions ![]()
transitions ![]() seulement.
seulement.
Valeurs
principales des tenseurs de couplage hyperfin des protons :
�������������� �����������![]() �(G)
�(G)
![]() �� ������2.5����������� 6.1������������ 4.3
�� ������2.5����������� 6.1������������ 4.3
![]() �������� �-6.4 ��������-18.9 ���������-12.5
�������� �-6.4 ��������-18.9 ���������-12.5
![]() �������� ���-5.8��������
-17.8��������� �-13.0
�������� ���-5.8��������
-17.8��������� �-13.0
Provenant
de C. Heller et T. Cole, J. Chem. Phys.
33, 243 (1962).
.Pour chaque proton l�axe principal Z de ces tenseurs est perpendiculaire au plan du radical, l�axe X �tant dirig� selon la liaison �C-H.
DISTRIB. �Correspond aux options 1 et 2 de RIGMAT, mais pour des
syst�mes partiellement orient�s :
-Distribution Gaussienne des
angles polaire et azimuthal de ![]() .
.
dans le syst�me XYZ des axes principaux des tenseurs a et
g
-Distribution gaussienne d�orientations autour d�un axe quelconque.
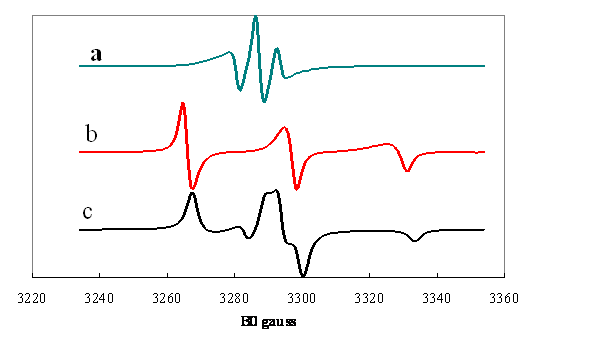
Figure 4. Spectres calcul�s
pour un radical nitroxyde :
a et b : distribution Gaussiennes des angles
polaire et azimuthal ![]() �de
�de ![]() �dans le r�f�rentiel
XYZ centr�es sur
�dans le r�f�rentiel
XYZ centr�es sur ![]() �(a),
�(a), ![]() �(b) pour un �cart-type
�(b) pour un �cart-type
![]() .
.
c : distribution sph�rique (isotrope) d�orientations.
MULTIP- Spectres d��tats triplets,
biradicaux et paires de radicaux ou d�ions en matrice rigide pour les
transitions ![]() �et
�et ![]() . Diff�rents programmes sont utilisables selon la sym�trie du
syst�me et l�existence ou non de couplages hyperfins. Dans le cas o� le tenseur
g, le tenseur d�interaction
dipolaire, et les tenseurs de couplage hyperfin n�ont pas les m�mes �orientations le spectre est calcul� suivant le
traitement par perturbation du second ordre d�Iwasaki.(6).
. Diff�rents programmes sont utilisables selon la sym�trie du
syst�me et l�existence ou non de couplages hyperfins. Dans le cas o� le tenseur
g, le tenseur d�interaction
dipolaire, et les tenseurs de couplage hyperfin n�ont pas les m�mes �orientations le spectre est calcul� suivant le
traitement par perturbation du second ordre d�Iwasaki.(6).
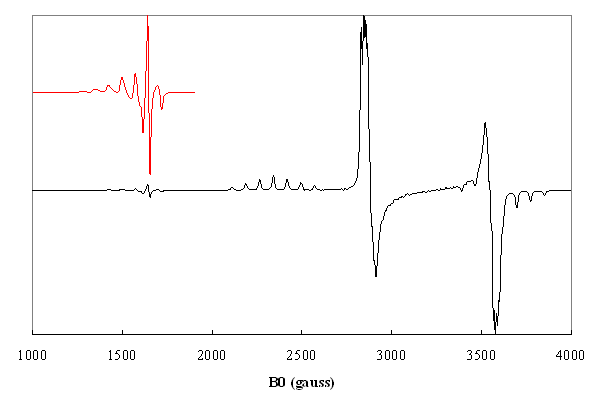
Figure5. Spectre d�une paire
d�ions ![]() dans l�oxyde CuCe calcul� pour les transitions
dans l�oxyde CuCe calcul� pour les transitions ![]() �avec les param�tres de
d�g�n�rescence en champ nul D =
�avec les param�tres de
d�g�n�rescence en champ nul D = ![]() ,
, ![]() (A.Aboukais,E. Abi-Aad, A. Bennani, C. Chachaty, J-P.
Bonnelle, J. Chem.Soc. Faraday Trans. 91, 3299 (1995))
(A.Aboukais,E. Abi-Aad, A. Bennani, C. Chachaty, J-P.
Bonnelle, J. Chem.Soc. Faraday Trans. 91, 3299 (1995))
BIRAD- Spectre d�un biradical
dont les groupes radicalaires sont reli�s par une cha�ne
alkyle. L�interaction d��change entre les deux spins �lectroniques varie
rapidement avec leur distance et d�pend du recouvrement des orbitales dans
lesquelles ils sont localis�s. Cette interaction modul�e par
l�isom�risation� trans��gauche autour des liaisons de la
cha�ne provoque une alternance des largeurs de raies. La simulation optimis�e
des spectres exp�rimentaux compl�t�e par des mesures des temps de relaxation T1 des
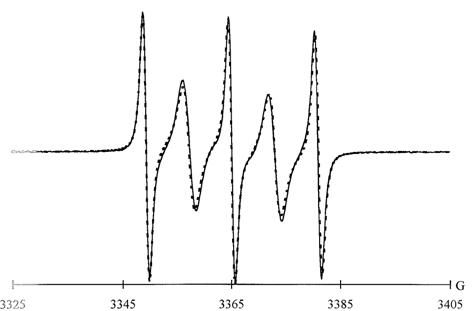
Figure 6. Spectres
exp�rimental (��) et calcul� (- - - - ) du biradical
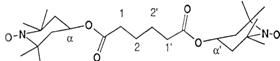
dans le t�trahydrofuranne � 353 K (C. Chachaty, S.
Gambarelli et A. Rassat, Magn. Res. in Chem 33, S174
(1995)).
HMLT- Lorsque le couplage hyperfin d�un noyau ou le couplage dipolaire
entre deux spins �lectroniques d�passe environ 10% de l�intensit� du champ
magn�tique directeur, le calcul de la position des raies de r�sonance par la
m�thode de perturbation du second ordre manque de pr�cision. Les valeurs
pr�cises de la position des raies de r�sonance sont donn�es par les transitions
entre les niveaux d��nergie du syst�me de spins calcul�s en fonction de
l�intensit� du champ magn�tique par diagonalisation de la matrice de l�Hamiltonien
de spin. Cette m�thode est appliqu�e ici aux spectres de radicaux libres en
phase liquide et aux �tats triplets ou aux biradicaux.
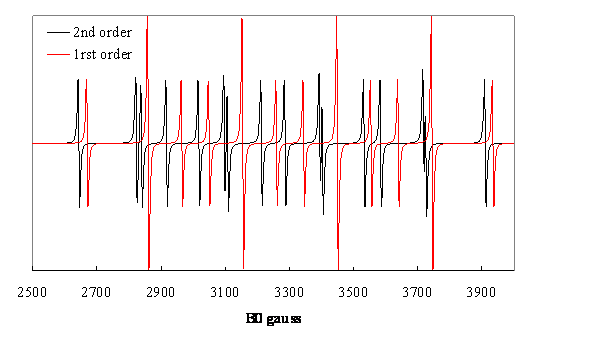
Figure 7. Spectres� de 1er et 2i�me� ordre de ![]() �calcul�s avec
�calcul�s avec ![]() ,
, ![]() ,� constantes de
couplages provenant de W. Nelson et W. Gordy, J. Chem. Phys. 51, 4710, (1969).
,� constantes de
couplages provenant de W. Nelson et W. Gordy, J. Chem. Phys. 51, 4710, (1969).
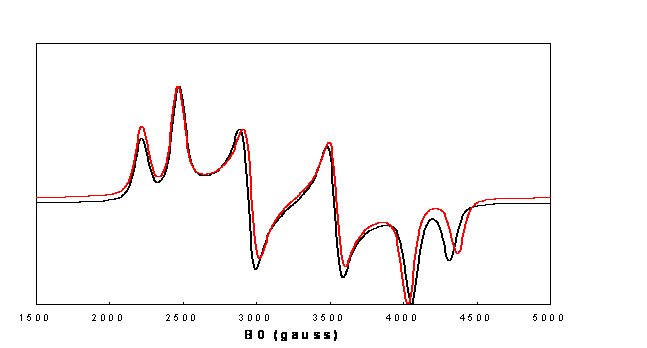
Figure 8.
Spectres de l��tat triplet du naphtal�ne polyorient�,
transition
DM=�1� calcul�s avec
D =
�� : Diagonalisation de la matrice de l�Hamiltonien de
spin, ����
perturbation du second ordre de l�Hamiltonien de spin(6).
![]() ) en milieux isotropes pour les cas suivants :
) en milieux isotropes pour les cas suivants :
-R�orientation rapide.
-R�orientation lente (diffusion brownienne) selon le
mod�le d��change multisites de McConnell et coll.(7).
-R�orientation lente avec distribution de temps de
corr�lation, pour les polym�res par exemple.
-Echange d�une sonde paramagn�tique entre un milieu
fluide et un milieu visqueux ou semi-rigide. La simulation optimis�e de
spectres exp�rimentaux donne en particulier les temps de corr�lation de
r�orientation, la vitesse d��change et la population des sites.
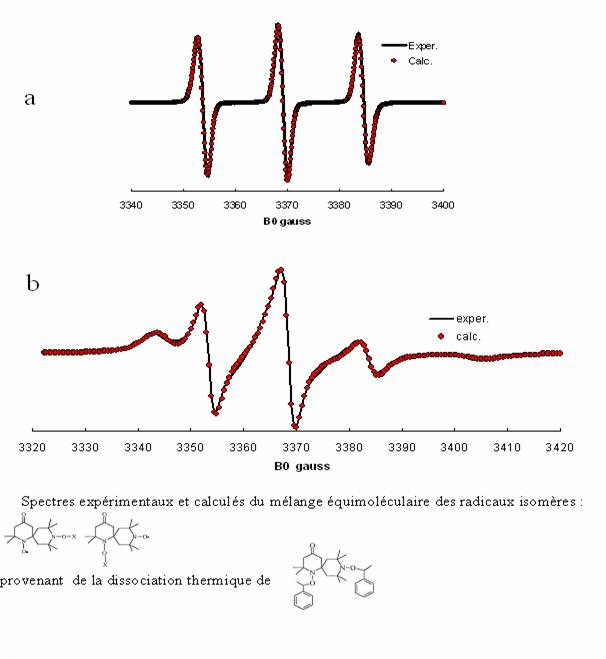
Figure 9. a
� Spectre initial des radicaux isom�res dans le styr�ne � 293 K, simul� pour ![]() ,
, ![]() et
et ![]() . X d�signe le groupe styryle.
. X d�signe le groupe styryle.
b � En cours de
polym�risation. Le groupe X est alors la cha�ne de polystyr�ne. La simulation
du spectre indique l�existence de deux fractions de temps de corr�lation de
r�orientation respectifs ![]() (18%) et
(18%) et ![]() �(C. Chachaty, Wenli
Huang, L. Marx, B. Charleux, A. Rassat, Polymer, 44, 397 (2003)).
�(C. Chachaty, Wenli
Huang, L. Marx, B. Charleux, A. Rassat, Polymer, 44, 397 (2003)).
FNJP
-R�orientation d�un radical par sauts d�amplitudes
finies.
-Echange d�un radical entre 2 ou 3 conformations.
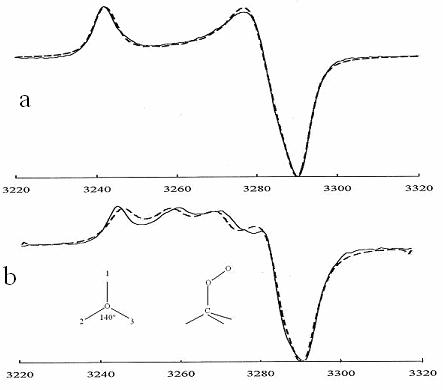
Figure 10. .Spectres
exp�rimentaux (��) et calcul�s (- - - - -) du radical peroxyle dans le
polypropyl�ne.
a � Vitesses de saut du
groupe O-O entre 3 orientations ![]()
b � ������������������ ���������������� ������� �����
![]() ,
,![]() ,
,
![]() .
.
LQCR- RPE de sonde paramagn�tiques dans les phases cristallines liquides anisotropes(1,2) pour les cas suivants:
-Phase
unique n�matique, lamellaire ou hexagonale.
-Echange
d�une sonde entre une phase anisotrope et une phase isotrope (liquide isotrope
ou phase cubique).
-Echange
entre deux sites anisotropes.
���� Pour une
r�orientation rapide de la sonde, la simulation automatis�e des spectres
exp�rimentaux donne principalement le param�tre d�ordre mol�culaire et les
temps de corr�lation de r�orientation de la sonde. Dans le cas d�un �change, on
obtient �galement la vitesse d��change et la population des sites. Lorsque la r�orientation
est lente on d�termine seulement les largeurs de raies et le� param�tre d�ordre mol�culaire que l�on peut
utiliser comme param�tres initiaux pour d�terminer les temps de corr�lation en
simulant les spectres avec le programme NLSL de Freed et coll.(8)
accessible depuis cette zone de travail dans la version APL+WIN.
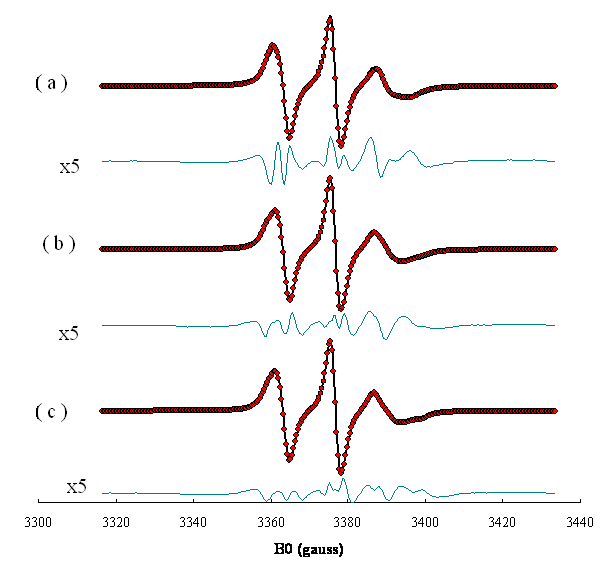
Figure 11-Spectres
exp�rimentaux (��), simul�s (◦
◦ ◦ ◦ ◦) et leurs diff�rences amplifi�es 5 fois (x5) de
la sonde �de� phosphatidylcholine marqu�e par un radical
doxyle en position 14 de la cha�ne sn2
(PC14) dans une bicouche de sphingomy�line�
contenant 17mol% de cholest�rol � ![]() �et du temps de correlation de r�orientation �
�et du temps de correlation de r�orientation �![]() �sont :�
�sont :�
( a )�� Dans l�hypoth�se
d�un seul site: ![]() �= 0.21,
�= 0.21, ![]() �=
0.95 ns.
�=
0.95 ns.
( b ) Pour 2 sites sans �change : fraction site I :
0.34, site I: ![]() = 0.33,
= 0.33, ![]() �= 0.59 ns, site II:
�= 0.59 ns, site II: ![]() = 0.14,
= 0.14, ![]() �= 1.0 ns
�= 1.0 ns
( c ) Pour 2 sites avec �change : fraction site I �0.52, vitesse d��change I↔II ![]() , site I :
, site I : ![]() = 0.36,
= 0.36, ![]() �= 0.45 ns, site II:
�= 0.45 ns, site II: ![]() = 0.14,
= 0.14, ![]() = 1.2 ns.
= 1.2 ns.
L��cart-type
entre le spectre exp�rimental� et les
spectres simul�s sont� (a) σ = 1%, (b) σ = 0.65%, (c)
σ = 0. 35%.
DOXYL- Simulation des spectres du
radical doxyle li� en diff�rentes position d�une
cha�ne flexible, par exemple la cha�ne sn2
d�un phospholipide dans la phase lamellaire d�une membrane
(9).
���� Les
param�tres ajustables sont le param�tre d�ordre mol�culaire et les temps de
corr�lation de r�orientation globale de la sonde, les populations du rotam�re trans autour des liaisons C-C et les
vitesses des mouvements segmentaires. Tous ces param�tres peuvent �tre obtenus
par simulation optimis�e des spectres prenant en compte les conform�res les
plus probables repr�sentant au moins 90% de leur population totale. Les
programmes fonctionnent selon les m�mes principes que ceux de la zone CHAINE d�crite plus loin.
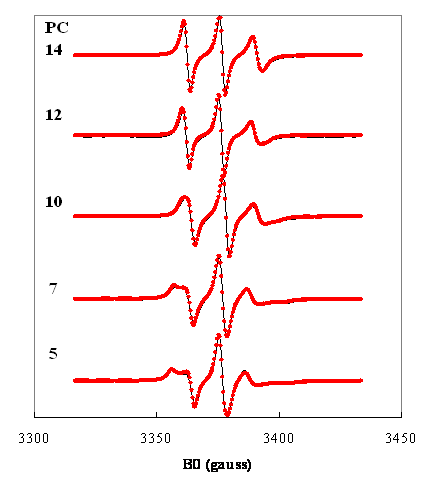
Figure 12. Spectres exp�rimentaux (��) et simul�s (������) de la
phosphatidylcholine (PC) marqu�e par un groupe doxyle� en positions 5 � 14 de la cha�ne sn2 dans une bicouche de sphingomy�line
� 326 K(9).
�
2-
R�sonance et relaxation nucl�aires
RMN1-Spectre RMN d�un noyau de spin quelconque en matrice rigide ou dans un cristal liquide. Les largeurs de raies sont ajust�es empiriquement ou calcul�es en fonction des temps de corr�lation de r�orientation mol�culaire(1).
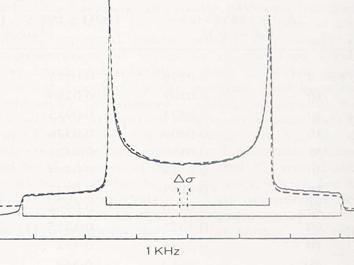
Figure
13. Spectres exp�rimental (��) et simul� (- - - - -)� de ![]() �de l�eau dans un
cristal liquide de dibutylphosphate/
�de l�eau dans un
cristal liquide de dibutylphosphate/![]() � 230 K. Param�tre d�ordre mol�culaire : 0.049,
anisotropie de d�placement chimique de
� 230 K. Param�tre d�ordre mol�culaire : 0.049,
anisotropie de d�placement chimique de ![]() :
: ![]() -1.37
ppm (C. Chachaty, J.P. Quaegebeur, Mol. Phys. 52, 1081 (1984)).
-1.37
ppm (C. Chachaty, J.P. Quaegebeur, Mol. Phys. 52, 1081 (1984)).
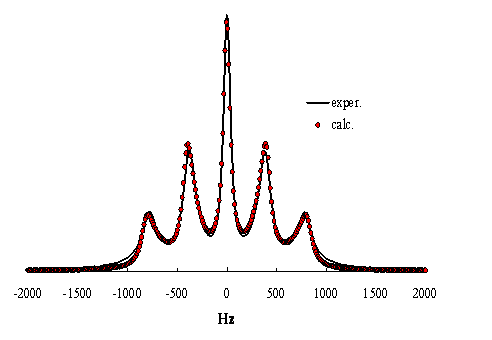
Figure
14. Spectre de l�oxyg�ne 17 de ![]() �adsorb� sur le DNA � 333
K . Le spectre exp�rimental provient de A. Catte,
Universit� de Cagliari, Italie. Les param�tres optimis�s par simulation de ce
spectre sont
�adsorb� sur le DNA � 333
K . Le spectre exp�rimental provient de A. Catte,
Universit� de Cagliari, Italie. Les param�tres optimis�s par simulation de ce
spectre sont ![]() �pour l�axe de sym�trie
mol�culaire dont le temps de corr�lation de r�orientation est
�pour l�axe de sym�trie
mol�culaire dont le temps de corr�lation de r�orientation est ![]()
RMN2- Spectre RMN d�un noyau de spin 1/2 ou 1 pour une r�orientation
mol�culaire lente en milieu isotrope. On utilise le mod�le de diffusion
brownienne ou de sauts al�atoires d�amplitudes�
finies comme pour les programmes des zones BRNDF et FNJP.
RELAX- Calcul des temps de
relaxation T1 et T2 d�un noyau de spin 1/2� ou 1 dans une mol�cule rigide en
r�orientation dans un liquide ou dans un segment de macromol�cule� flexible.�
Dans ce dernier cas, on peut choisir diff�rentes fonctions de
distribution des temps de corr�lation de r�orientation.
CHAINE- Relaxation dipolaire ou
quadrupolaire des carbones 13 , des protons ou des
deut�rons d�une cha�ne alkyle li�e � une t�te polaire (par ex. �groupes ![]() ,
, ![]() �ou
�ou ![]() ) en r�orientation dans un liquide ou dans un cristal liquide(4,10).
) en r�orientation dans un liquide ou dans un cristal liquide(4,10).
-Ecarts dipolaires ou quadrupolaires des raies de ces m�mes noyaux dans un cristal liquide(10).
-Relaxation dipolaire ou d�placement de
pseudocontact des carbones 13 ou des protons de la cha�ne induits par un ion
paramagn�tique fix� � la t�te polaire(3, 11).
-Relaxation dipolaire d�un noyau de spin 1/2 de la
t�te polaire (par ex. phosphore ou azote 15) par les protons de la cha�ne
alkyle.
���� Les calculs de ces observables sont bas�s sur le mod�le de l�isom�risation trans��gauche autour de chaque liaison C-C de la cha�ne.
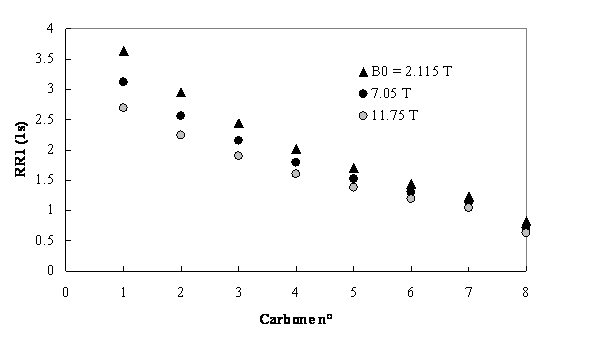
Figure 15. Vitesses de relaxation longitudinale des
carbones 13 de la cha�ne alkyle de l�octylphosphate de pyridinium en phase
lamellaire � 300 K. Ces vitesses sont calcul�es d�apr�s les donn�es de la r�f.
(10) avec un param�tre d�ordre ![]() �et des temps de
corr�lation de r�orientation mol�culaire
�et des temps de
corr�lation de r�orientation mol�culaire![]() . La population du rotam�re trans� autour des liaisons
C-C est 0.9 et la probabilit� par unit� de temps de la transition gauche→trans est
. La population du rotam�re trans� autour des liaisons
C-C est 0.9 et la probabilit� par unit� de temps de la transition gauche→trans est
![]() .
.
3-Ex�cution d�un programme.
-Charger
la zone de travail correspondant au probl�me � traiter
-Entrer
en minuscules le nom de la zone pour avoir la liste des programmes avec leurs
caract�ristiques. Ceux-ci existent g�n�ralement sous deux formes : simple
simulation pour �tudier l�influence des param�tres spectroscopiques et/ou
dynamiques sur la forme des spectres ou d�termination des valeurs optimis�es de
ces param�tres par simulation automatis�e d�un spectre exp�rimental.
�-Entrer le nom du programme choisi. Celui-ci
appelle g�n�ralement un sous-programme pour introduire ou modifier les
param�tres, autrement cela est effectu� par �tapes au cours de l�ex�cution du
programme principal.
Exemple :
(les commentaires non inclus dans le programme sont inscrits
en rouge entre crochets.)
)LOAD RIGMAT������ [Charger la zone (workspace)]
SAVED 2005-12-23 13.58.02
(GMT-5)
NONAX1F [Afficher le nom du programme choisi puis ↓ Entr�e]
Is
the spectrum a DOS file ? [1]
�:
����� 0��������������������
Name of the spectrum :
�:
����� RTM125 [le
tableau XY correspondant existe d�j� dans la zone]
Number
of points :� 528
Baseline
correction ? [1]
�:
����� 0����������������� [ligne
de base d�j� rectifi�e]
Normalized
spectrum : XY, integral : IXY
Second
moment (M2) :�
60048.8
Hm for g = g0 :� 3248 [Hm donne approx.le centro�de du spectre]
Spectral range about Hm :
-669 912
NONAX1F_IN�� [cliquer sur ce nom pour �diter les param�tres initiaux et les modifier �ventuellement]

[Terminer
l��dition/correction des donn�es par ↓ cl�
F4 puis ↓� Entr�e.Les donn�es initiales sont
r�capitul�es ci-dessous]
gZZ, gYY, gXX :� 1.9375 1.973 1.974
g0 :� 1.9615
Field
H0 g = 2.00232 :�
3181.78
Frequency : 8.91681 GHz
Correction to H0? : DH0≠0 : 35
Field
Hc for g0 = 1.9615 :�
3248
Total
nuclear spin :�
3.5
Relative
line intensities :�
1 1 1 1 1 1 1 1
aZZ,aYY,aXX (a,gauss) :� 185 71 66
a0 =� 107.333
Second
order ? [1] : 1
Angular
range of θ:� 0 90
Uneven
number of orientations�
91
Standard dev. of Gaussian distrib. of θ (dth≠0) : 0
Lorentzian
[1] or Gaussian [2] lineshape ?� 1
Linewidths� ∆HRzz, ∆HRyy,
∆Hxx :� 5 5 5
Stand.
dev. of the Gaussian broadening
or half width of the Lorentz. broadening
(∆HGL) :� 3
Integral ? [1] :� 0
Automated fit?[1]� [si 0 simulation pr�alable du spectre pour bien centrer le
spectre calcul�/spectre exp�rimental et v�rifier la validit� des donn�es
initiales]
�:
����� 1
Iteration max. number :
�:
����� 120
[Ecart-type
normalis� entre spectre exper. et spectre calc., vs
nombre d�it�rations]
10 13.6243
20
12.6999
30
9.84888
40
3.92905
50
3.94236
60
2.44939
70
2.30148
80
2.27829
90
2.28054
100
2.26737
110
2.26128
120
2.25777
130 2.25776

More iterations ? :
�:
����� 0 [si
l�optimisation ne progresse plus ignificativement]
Name
of the XY1..N table
SP1���� [donner un nom aux
spectres exp. et calc. pour les conserver dans 1a zone ou les tracer
ult�rieurement sur Excel]
Iteration
number :� 131
Calc.
time (s) :�
399.593
Input
parameters [1], manual↔automated fit [2]
Optimized
parameters [3], replot [4], Excel [5]
�:
����� 3
Title?
RTM125
NONAX1F
* RTM125 *� 23
12 2005
Standard
deviation![]() 100 :� 2.2556
100 :� 2.2556
H0
(g = 2.0023) :�
3216.8
gzz, gyy, gxx and g0 :� 1.9377 1.9732
1.9742�� 1.9617
Nuclear
spin :� 3.5
azz, ayy, axx and a0 :� 184.19 68.856
64.368�� 105.8
Spectral
window and resolution :� 2579 4160 3
∆HRzz, ∆HRyy, ∆Hxx Lorentz. :� 6.9886 3.2892 7.936
Gaussian broadening : 3.7668
[convolution du spectre calcul� par une Gaussienne]
Input
parameters [1], manual↔automated fit [2]
Optimized
parameters [3], replot [4], Excel [5]
�:
����� 0
---------------------------------------------------------------Another
spectrum ? [1]
�:
����� 0
4 � R�f�rences
4. J.Ph Caniparoli, A.
Grassi, C. Chachaty.. Mol.. Phys. 63, 419 (1988).
�� b. D.W. Marquardt,� J.
Soc. Ind. Appl. Math. 11,
431 (1963).
6�
M. Iwasaki, J. Mag. Res. 16, 417
(1974).
7. R.C. McCalley,
E.J. Shimshick, H.M. McConnell, Chem. Phys. Letters, 13, 115 (1972).
8. D.E. Budil, S. Lee, S. Saxena, J.H.
Freed. J. Magn. Res. Series A. 120,: 155 (1996).