UNE APPLICATION D’APL EN MECANIQUE QUANTIQUE
: LES NIVEAUX D’ENERGIE D’UN SYSTEME DE SPINS ELECTRONIQUES ET NUCLEAIRES
Claude Chachaty
claude.chachaty@wanadoo.fr
Dans un
précédent article(1), j’ai donné comme exemple d’application scientifique
d’APL, la simulation des spectres de résonance paramagnétique électronique
(RPE). Cette branche de la spectroscopie hertzienne permet d’identifier et de
doser les molécules paramagnétiques qui interviennent dans une multitude de
réactions chimiques, en particulier la combustion et la photosynthèse. Les
molécules paramagnétiques, dont l’oxygène de l’air fait partie, comportent un
ou plusieurs électrons non appariés qui possèdent un spin ![]() ,
auquel est associé un moment magnétique
,
auquel est associé un moment magnétique ![]() où
ge est le facteur spectroscopique électronique voisin de 2 pour l’électron
libre et be , le magnéton de Bohr.
où
ge est le facteur spectroscopique électronique voisin de 2 pour l’électron
libre et be , le magnéton de Bohr.
Dans un champ magnétique ![]() ,
, ![]() et
et ![]() prennent
2S+1 orientations auxquelles correspondent autant de niveaux d’énergie, c’est
l’effet Zeeman bien connu des spectroscopistes. S est le nombre quantique de
spin, multiple de 1/2, qui pour n électrons non appariés est égal à n/2.
Chaque orientation est caractérisée par la projection de
prennent
2S+1 orientations auxquelles correspondent autant de niveaux d’énergie, c’est
l’effet Zeeman bien connu des spectroscopistes. S est le nombre quantique de
spin, multiple de 1/2, qui pour n électrons non appariés est égal à n/2.
Chaque orientation est caractérisée par la projection de ![]() sur
sur ![]() ,
égale à l’une des valeurs du nombre quantique électronique
,
égale à l’une des valeurs du nombre quantique électronique ![]() .
Selon le principe d’exclusion de Pauli, une orbitale atomique ou moléculaire ne
peut compter que deux électrons de spins opposés, c’est à dire dans les états
.
Selon le principe d’exclusion de Pauli, une orbitale atomique ou moléculaire ne
peut compter que deux électrons de spins opposés, c’est à dire dans les états ![]() et
et ![]() ,
ou un seul électron (électron non apparié) dans l’un de ces deux états.
,
ou un seul électron (électron non apparié) dans l’un de ces deux états.
Les
noyaux atomiques possèdent pour la plupart un spin ![]() ,
auquel est associé un moment magnétique
,
auquel est associé un moment magnétique ![]() où
où ![]() est
le facteur spectroscopique spécifique de chaque noyau et
est
le facteur spectroscopique spécifique de chaque noyau et ![]() ,
le magnéton nucléaire, inférieur par trois ordres de grandeur au magnéton de
Bohr. A l’exception évidente du principe d’exclusion, tout ce qui est dit plus
haut pour le spin électronique est valable pour les spins nucléaires en
remplaçant S par I et
,
le magnéton nucléaire, inférieur par trois ordres de grandeur au magnéton de
Bohr. A l’exception évidente du principe d’exclusion, tout ce qui est dit plus
haut pour le spin électronique est valable pour les spins nucléaires en
remplaçant S par I et ![]() par
par ![]() .
I = 1/2 pour le proton, le carbone 13, l’azote 15 et le phosphore 31 , I = 1
pour le deutéron et l’azote 14, I = 5/2 pour l’oxygène 17, par
contre le carbone 12 et l’oxygène 16 n’ont pas de spin ( I = 0 ).
.
I = 1/2 pour le proton, le carbone 13, l’azote 15 et le phosphore 31 , I = 1
pour le deutéron et l’azote 14, I = 5/2 pour l’oxygène 17, par
contre le carbone 12 et l’oxygène 16 n’ont pas de spin ( I = 0 ).
Pour simplifier, nous ne considérons ici que l’exemple d’un électron non
apparié (S = 1/2) soumis à des interactions magnétiques isotropes avec un champ
continu d’intensité B et un noyau de spin I. C’est le cas par exemple
d’un atome d’hydrogène en phase gazeuse ou d’un ion métallique tel que ![]() en
phase liquide, pour lesquels les interactions anisotropes sont soit
inexistantes, soit annulées en moyenne.
en
phase liquide, pour lesquels les interactions anisotropes sont soit
inexistantes, soit annulées en moyenne.
L’interaction du spin électronique S = 1/2 avec le champ magnétique directeur ![]() donne
deux niveaux d’énergie
donne
deux niveaux d’énergie ![]() .
La transition entre ces niveaux induite par un photon d’énergie
.
La transition entre ces niveaux induite par un photon d’énergie ![]() ,
où n est la fréquence du spectromètre et h, la constante de Planck, donne lieu
à une raie unique d’absorption observable par RPE. Lorsque le spin électronique
interagit avec un spin nucléaire qui exerce sur lui un champ magnétique
local dépendant de
,
où n est la fréquence du spectromètre et h, la constante de Planck, donne lieu
à une raie unique d’absorption observable par RPE. Lorsque le spin électronique
interagit avec un spin nucléaire qui exerce sur lui un champ magnétique
local dépendant de ![]() ,
les deux niveaux fondamentaux d’énergie se divisent en 2I+1 sous-niveaux. On
obtient alors 2I+1 raies de résonance correspondant aux transitions permises
,
les deux niveaux fondamentaux d’énergie se divisent en 2I+1 sous-niveaux. On
obtient alors 2I+1 raies de résonance correspondant aux transitions permises ![]() .
La multiplicité des raies de résonance et leurs écarts permettent d’identifier
l’espèce paramagnétique observée par RPE et donnent des informations sur la
configuration électronique et la conformation moléculaire. Des règles simples
permettent de calculer les positions approximatives des raies de
résonance et de chercher leur meilleur accord avec l’expérience.
L’approximation est d’autant meilleure que la fréquence et donc le champ
magnétique du spectromètre est plus élevé, mais il est parfois nécessaire de
travailler en champ faible ce qui nécessite, comme on le verra, de disposer
d’une méthode rigoureuse de calcul. Cette méthode consiste à rechercher les
fonctions de spin
.
La multiplicité des raies de résonance et leurs écarts permettent d’identifier
l’espèce paramagnétique observée par RPE et donnent des informations sur la
configuration électronique et la conformation moléculaire. Des règles simples
permettent de calculer les positions approximatives des raies de
résonance et de chercher leur meilleur accord avec l’expérience.
L’approximation est d’autant meilleure que la fréquence et donc le champ
magnétique du spectromètre est plus élevé, mais il est parfois nécessaire de
travailler en champ faible ce qui nécessite, comme on le verra, de disposer
d’une méthode rigoureuse de calcul. Cette méthode consiste à rechercher les
fonctions de spin ![]() qui
vérifient l’équation :
qui
vérifient l’équation :
![]() [1]
[1]
H est
l’opérateur hamiltonien de spin et E sont les énergies correspondant aux
fonctions ![]() .
Celles-ci sont des combinaisons linéaires des (2S+1)(2I+1) fonctions de spin
.
Celles-ci sont des combinaisons linéaires des (2S+1)(2I+1) fonctions de spin ![]() qui
constituent tous les états possibles du système spin électronique, spin
nucléaire que nous représenterons par le vecteur de vecteurs
qui
constituent tous les états possibles du système spin électronique, spin
nucléaire que nous représenterons par le vecteur de vecteurs ![]() .
Les composantes
.
Les composantes ![]() sont
obtenues en APL étendu par l’opération MSMI„,MS°.,MI qui donne par exemple pour
S = 1/2 (
sont
obtenues en APL étendu par l’opération MSMI„,MS°.,MI qui donne par exemple pour
S = 1/2 (![]() )
et I = 1 (
)
et I = 1 (![]() 1, 0, -1) :
1, 0, -1) :
|MS,MI> = 0.5 1 0.5 0 0.5 ¯1 ¯0.5 1 ¯0.5 0 ¯0.5 ¯1
L’hamiltonien de spin est la somme de deux termes, l’un représentant
l’interaction entre les spins et le champ ![]() (terme
Zeeman) , l’autre correspondant au couplage scalaire entre les spins (couplage
hyperfin)(2, 3) . Dans un référentiel X,Y,Z dont l’axe Z est aligné sur
(terme
Zeeman) , l’autre correspondant au couplage scalaire entre les spins (couplage
hyperfin)(2, 3) . Dans un référentiel X,Y,Z dont l’axe Z est aligné sur ![]() ,
il a pour expression :
,
il a pour expression :
![]() [2]
[2]
a est la constante de
couplage hyperfin. Les composantes SX,Y,Z et I X,Y,Z de ![]() et
et ![]() sont
des opérateurs de spin qui agissent indépendamment sur les composantes
sont
des opérateurs de spin qui agissent indépendamment sur les composantes ![]() et
et ![]() des
fonctions de spin
des
fonctions de spin ![]() .
Nous ne donnons que ceux relatifs au spin nucléaire, utilisés ici :
.
Nous ne donnons que ceux relatifs au spin nucléaire, utilisés ici :
![]() [3]
[3]
![]() [4]
[4]
![]() [5]
[5]
Les
opérateurs ![]() et
et ![]() augmentent
et diminuent respectivement |
augmentent
et diminuent respectivement |![]() >
d’une unité selon que
>
d’une unité selon que ![]() <
I ou
<
I ou ![]() >
-I et annihilent respectivement
>
-I et annihilent respectivement ![]() et
et ![]() .
.
On
obtient des relations analogues pour le spin électronique en changeant I en
S, ![]() en
en ![]() etc...
, de sorte que l’on peut remplacer
etc...
, de sorte que l’on peut remplacer ![]() par
par ![]() dans
la relation [2]. Le résultat des opérations
dans
la relation [2]. Le résultat des opérations ![]() est
le produit des opérations respectives de
est
le produit des opérations respectives de ![]() et
de
et
de ![]() sur
sur ![]() et
et ![]() .
Il en est de même pour
.
Il en est de même pour ![]() .
.
Les niveaux d’énergie entre lesquels s’effectuent les transitions sont les valeurs propres de la matrice de l’hamiltonien de spin dont les éléments sont
![]() et
que nous appellerons matrice H. Les éléments diagonaux de cette matrice sont
obtenus par action sur les fonctions de spin, des termes contenant
et
que nous appellerons matrice H. Les éléments diagonaux de cette matrice sont
obtenus par action sur les fonctions de spin, des termes contenant ![]() et
et ![]() qui
laissent inchangées les fonctions de spin (relation 3). Les éléments non
diagonaux proviennent du terme
qui
laissent inchangées les fonctions de spin (relation 3). Les éléments non
diagonaux proviennent du terme ![]() qui
modifie ou annule les fonctions de spin (relation 5) :
qui
modifie ou annule les fonctions de spin (relation 5) :
![]() [6 ]
[6 ]
![]() [7 ]
[7 ]
Pour construire
la matrice des éléments non diagonaux de H, on crée d’abord la matrice binaire
qui compare les vecteurs composant ![]() à
ceux de
à
ceux de ![]() et
de
et
de ![]() au
moyen de l’opérateur APL °.=
comme l’indique la fonction :
au
moyen de l’opérateur APL °.=
comme l’indique la fonction :
NDIAG„hmlt SN
©
© - SN est le spin nucléaire
© Vecteur de vecteurs |MS,MI>:
©
MS„0.5 ¯0.5 ª MSMI„,MS°.,MI„SN,SN-¼2×SN
©
© - L'opérateur S(+)I(-) augmente MS et diminue MI de 1
©
MSP„MS+1 ª MIM„MI-1
©
© - Vecteur de vecteurs |MS+1,MI-1>:
©
SPIM„,MSP°.,MIM
©
© - Les composantes "interdites" de |MS+1,MI-1> sont
© - éliminées par l'opération suivante.
© - Eléments communs à |MS,MI> et à |MS+1,MI-1>:
©
NDIAG„×/¨MSMI°.=SPIM
©
© - L'opérateur S(-)I(+) ayant une action symétrique
© - de celle de S(+)S(-) sur |MS,MI> , il est
© - inutile de répéter les 2 opérations précédentes.
©
NDIAG„NDIAG+³NDIAG
Les éléments
non diagonaux de la matrice H sont obtenus en multipliant NDIAG par a/2 et par
les coefficients ![]() appropriés,
désignés par k’ et k ’’ dans les relations 6 et 7. Un exemple de
matrice H est donné dans le Tableau I pour le système
appropriés,
désignés par k’ et k ’’ dans les relations 6 et 7. Un exemple de
matrice H est donné dans le Tableau I pour le système ![]() I
= 1.
I
= 1.
On remarque dans ce tableau, que les éléments diagonaux sont fonction de l’intensité du champ magnétique B, que l’on fait varier linéairement pour enregistrer un spectre de RPE à fréquence fixe n0. Pour calculer les positions des raies de résonance, il faut déterminer les valeurs propres E de la matrice H pour chaque valeur de B puis résoudre en fonction de ce paramètre, les équations :
![]() [8]
[8]
ou inversement, en fonction
de n si l’on travaille à champ fixe et à fréquence variable. Ei et Ej sont deux
niveaux d’énergie vérifiant la règle de sélection ![]() .
La fonction SYMEIG d’APL*PLUS utilisée pour diagonaliser H a le défaut de
donner dans n’importe quel ordre les valeurs propres qu’il faut trier ensuite
pour obtenir des fonctions E(B) continues.
.
La fonction SYMEIG d’APL*PLUS utilisée pour diagonaliser H a le défaut de
donner dans n’importe quel ordre les valeurs propres qu’il faut trier ensuite
pour obtenir des fonctions E(B) continues.
En champ élevé, quand les termes non diagonaux de H deviennent négligeables devant les termes diagonaux, les niveaux d’énergie sont donnés au premier ordre par ceux-ci et l’on obtient 2I+1 raies équidistantes pour des champs magnétiques:
![]() [9]
[9]
Lorsque
a est inférieur à ![]() d’un
ordre de grandeur seulement, la position des raies est donné avec une bonne
précision par un calcul de perturbation(2, 3). Si par contre a et
d’un
ordre de grandeur seulement, la position des raies est donné avec une bonne
précision par un calcul de perturbation(2, 3). Si par contre a et ![]() sont
du même ordre comme dans l’exemple choisi pour les figures 1 et 2, cette
détermination nécessite la diagonalisation de H.* (Dans ce tableau ,
sont
du même ordre comme dans l’exemple choisi pour les figures 1 et 2, cette
détermination nécessite la diagonalisation de H.* (Dans ce tableau , ![]() désignent
respectivement
désignent
respectivement ![]() .)
.)
|
|
1/2, 1 (I) |
1/2, -1 (III) |
-1/2,
1 (IV) |
-1/2,
0(V) |
-1/2,
-1 (VI) |
|
1/2, 1 (I) |
|
0 |
0 |
0 |
0 |
|
1/2, 0 (II) |
0 |
0 |
|
0 |
0 |
|
1/2, -1 (III) |
0 |
|
0 |
|
0 |
|
-1/2, 1 (IV) |
0 |
0 |
|
0 |
0 |
|
-1/2, 0 (V) |
0 |
|
0 |
|
0 |
|
-1/2, -1 (VI) |
0 |
0 |
0 |
0 |
|
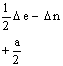
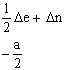
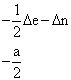
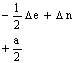
Tableau I. Matrice de l’hamiltonien de spin pour S = 1/2, I = 1(*)
|
|
||
|
Figure 1. Transitions entre les niveaux d’énergie d’un système S =
1/2, I = 1 |
||
|
|
||
|
|
|
|
|
Figure 2. Spectre d’un radical nitroxyde à 100 MHz (dérivée
première) Positions exactes des raies de résonances
: 15.84 27.37 47.29 G Calculées avec l’approximation du 1er ordre :
19.94 34.94 49.94 G (- - - - - -) Différence: 4.10
7.57 2.65 G |
|

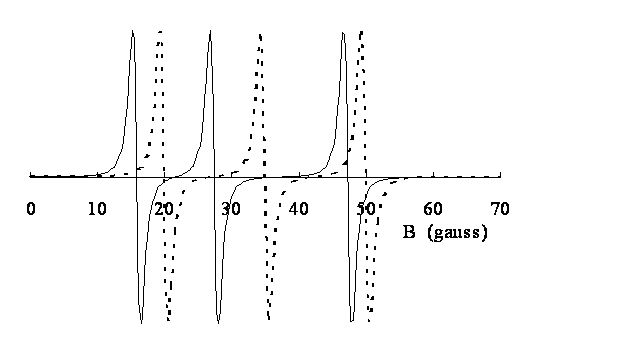
Les méthodes classiques d’interprétation des spectres de RPE utilisent l’approximation du premier ordre, ce qui revient à négliger implicitement les termes non diagonaux de H. La figure 2 montre l’importance des erreurs que cette approximation introduit dans l’exploitation des mesures effectuées à champ faible, en magnétométrie par exemple(4). De telles erreurs peuvent également se produire lorsque le couplage hyperfin dépasse une centaine de gauss, pour des mesures effectuées aux fréquences usuelles de 9-10 GHz, avec des champs magnétiques de l’ordre de 3000 à 3500 G.
La fonction permettant de donner les niveaux d’énergie et de calculer les
spectres pour un nombre quelconque de noyaux est écrite en APL*PLUS II et
APL*PLUS III. Une limite pratique d’utilisation de cette fonction est le
temps nécessaire pour diagonaliser en série plusieurs centaines de matrices
dont la taille est ![]() .
Il est possible de réduire le temps de calcul en assimilant un groupe de n
noyaux équivalents à un seul, de spin fictif nI et en limitant la
méthode de diagonalisation aux noyaux dont les constantes de couplage sont, par
exemple, supérieures à 0.1hn0, les couplages des autres étant traités par
l’approximation du premier ordre. Les positions Br de toutes les raies de
résonance sont alors données par l’expression APL, facile à généraliser :
.
Il est possible de réduire le temps de calcul en assimilant un groupe de n
noyaux équivalents à un seul, de spin fictif nI et en limitant la
méthode de diagonalisation aux noyaux dont les constantes de couplage sont, par
exemple, supérieures à 0.1hn0, les couplages des autres étant traités par
l’approximation du premier ordre. Les positions Br de toutes les raies de
résonance sont alors données par l’expression APL, facile à généraliser :
Br„,Bh°.-a1×MI1 ª Br„,Br°.-a2×MI2 etc...
Bh étant les champs de résonance obtenus par diagonalisation de H, a1, a2... , les constantes de couplage intervenant dans le traitement au premier ordre et MI1, MI2, les nombres quantiques nucléaires correspondants.
Ces exemples montrent qu’APL, parce qu’il est bien adapté au calcul vectoriel et matriciel, est un remarquable outil de recherche en physique fondamentale, en particulier pour la mécanique quantique et la spectroscopie moléculaire.
Références
1 - C. Chachaty, Les Nouvelles d’APL, n° 15, Mai 1995, pp 18-26.
2 - A. Carrington et A.D. McLachlan,
Introduction to Magnetic Resonance, Harper & Row, New-York 1967.
3 - W. Gordy, Theory and Applications of
Electron Spin Resonance,
John Wiley & Sons, New-York 1980.
4 - D. Djiret, M. Beranger, A. Bernerd, C. Jandey et M. Moussavi,
J. Chim. Phys., 91, 1862 - 1867 (1994).